小药说药新春第三谈:抗体偶联药物
前言
抗体偶联药物(ADC)是由靶向特异性抗原的单克隆抗体与小分子细胞毒性药物通过连接子链接而成,兼具传统小分子化疗的强大杀伤效应及抗体药物的肿瘤靶向性。ADC由三个主要部分组成:负责选择性识别癌细胞表面抗原的抗体,负责杀死癌细胞的药物有效载荷,以及连接抗体和有效载荷的连接子。
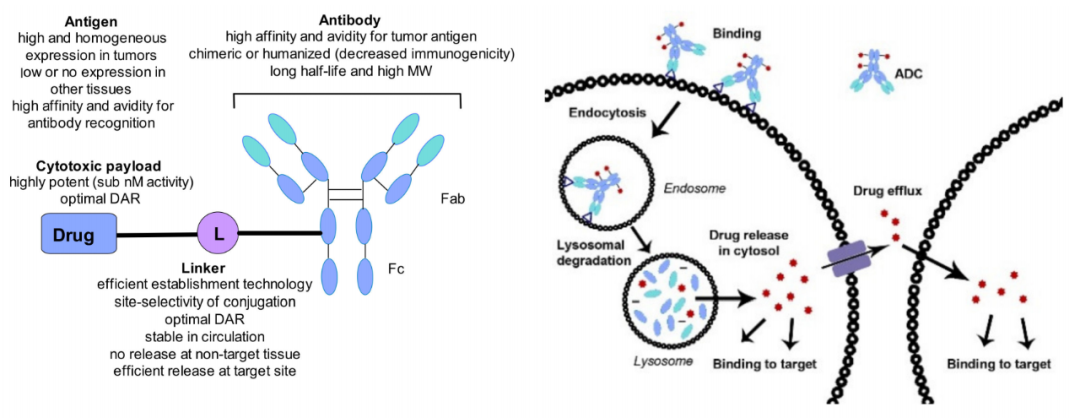
ADC对抗原的识别导致ADC通过内吞途径进入细胞内,通过溶酶体降解后,有效载荷以生物活性形式释放并发挥作用,导致癌细胞死亡。细胞内有效载荷的数量由每个细胞表面抗原的数量、每个ADC的药物有效载荷分子的数量(也称为药物抗体比率,DAR)以及抗原返回细胞表面所需的时间决定。有效载荷可能在癌细胞死亡和降解后逃逸,也可能从胞浆中透膜而出。这种释放的后果可能是有益的(也称为旁观者效应),也可能是有害的,导致全身毒性。
第一个ADC(Mylotarg)于2000年获得批准,自2019年以来,获准的ADC数量翻了一番多,2019-2020年共有5个ADC获得批准,ADC领域持续火热。
ADC药物的基本原理
ADCs药物的靶向性来自其中抗体部分,毒性大部分来自小分子化药毒物部分(payload),抗体部分也可以自带毒性(ADCC与CDC)。抗体部分与毒素部分通过连接子(linker)互相连接。抗体部分与肿瘤细胞表面的靶向抗原结合后,肿瘤细胞会将ADC内吞。之后ADC药物会在溶酶体中分解,释放出活性的化药毒物,破坏DNA或阻止肿瘤细胞分裂,起到杀死细胞的作用。理想化的连接子应该保持稳定所以不会导致靶外毒性(off-target toxicity),并且在细胞内高效释放毒物。

一个理想的ADC药物需要四个部分的完美结合:抗体(靶点)的选择:选择中需要考虑几个因素。
首先,理想的目标抗原在肿瘤中具有高表达水平,在正常组织中很少或没有表达,或至少表达限于给定组织类型,以减少ADC靶内毒性及导致可接受的治疗指数。
第二,目标抗原应存在于细胞表面,以便循环的mAb可进入。
第三,目标抗原应该是内在抗原,使其在结合后,ADC被转运到细胞中,细胞毒性剂可以发挥其作用。
连接子的选择:循环中药物过早释放可导致全身毒性和较低的治疗指标。有效的连接子设计必须平衡在循环中的几天内的良好稳定性以及在递送到靶细胞时的高效裂解。
对应的几种策略:目标细胞的细胞质中药物的有条件释放(可裂解和不可裂解的连接子);旁观者效应的增强和限制,是通过是否能够跨越生物膜的连接子-药物代谢产物实现的;极性连接子提高溶解度并降低MDR。
连接方式:第二代ADC均为不同药物负载物质的受控混合物,典型平均DAR为3.5或4。DAR大于4的物种显示较低的耐受性,更高的血浆清除率和降低的体内功效。目前大多数ADC具有共同的结构特征,如通过硫醇和烷基的马来酰亚胺反应形成的硫代琥珀酰亚胺连接。但大多数ADC长时间流通期间会导致可测量的马来酰亚胺消除,这些可通过位点特异性偶联和替代共轭化学来解决:如工程半胱氨酸、非天然氨基酸工程、酶辅助连接、糖重组和糖结合、氨基末端工程丝氨酸、与Fab核苷酸结合位点连接、天然半胱氨酸再桥接、避免retro-Michael 解体以及高负载ADC。
毒素分子:通过抑制微管蛋白装配而起作用的auristatin和美登木素生成素是目前ADC最常用的弹头。其他使用的弹头基于吡咯并苯并二氮杂(PBD)、二氢吲哚并噻二氮杂、Tubulysins、卡奇霉素、伊立替康衍生物、多卡莫霉素、喜树碱类类似物、肾上腺素和多柔比星等等。事实上,由于竞争激烈,在早期临床试验研究中越来越多的ADC,没有披露抗原靶和/或弹头和连接子的化学结构。
ADC的发展史
抗体药物的研发技术一直在不断更新换代,ADC的研究可以追溯到1980年,但是直到2000年,首个抗体偶联药物才被FDA批准用于治疗急性粒细胞白血病,但由于致死性的毒性的产生,于2010年撤市。随着原有技术的改进,研究人员开发了新型抗体偶联药物,并于2011年被FDA批准用于治疗霍奇金淋巴瘤和系统性间变性大细胞淋巴瘤。2013年抗体偶联药物再次取得突破,Genentech/ImmunoGen联合开发的Ado-trastuzumab emtansine被FDA批准用于HER2阳性乳腺癌,这是首个针对实体瘤的抗体偶联药物。随着这两个药物的研发成功,ADC药物再次以火热的状态进入人们的研究视野。
第一代抗体偶联药物—Mylotarg
像所有新技术发展的路径一样,大多数的第一代的产品因为有很多缺陷成了炮灰。在批准后一项研究中,Mylotarg与化疗相结合并未表现出比单独化疗更高的生存率和更高的致死毒性率,这导致辉瑞公司2010年自愿将此该药物从市场上撤出。第一代ADC药物失败的原因有很多因素,首先就是药物效力不足,血液中药物浓度低于治疗有效浓度,而靶点抗原低表达又导致药物递送量少,细胞内药物不足以杀死细胞。其次,初代ADC药物对肿瘤的靶向性不强,定位率低,而当时使用的连接子也不稳定,以致于药物毒性较大。最后,由于早期ADC中单克隆抗体是鼠源而不是人源化抗体,导致免疫反应和人抗鼠抗体(HAMAs)的产生。以上都是第一代药物失败的因素,但是研究永无止境,很快第二代ADC药物进入人们的视野。

Mylotarg(Gemtuzumab ozogamicin)结构式
第二代抗体偶联药物—Kadcyla
罗氏的Trastuzumab emtansine(Kadcyla)于2013年2月获得FDA批准,用于治疗之前接受过曲妥珠单抗和紫杉类化疗失败的HER2阳性乳腺癌患者。Kadcyla,是一个靶向HER2 抗体药物偶联物,含人源化抗HER2 IgG1曲妥珠单抗(Trastuzumab)和微管抑制剂DM1(美登素maytansine衍生物),两者通过稳定硫醚连接物(MCC)共价连接。第二代ADC药物的研发中,mAb技术得到改进,单克隆抗体被仔细选择,提高了肿瘤细胞靶向性,并减少与健康细胞交叉反应。更重要的是,早期使用当时治疗癌症的小分子药物作为毒性荷载缺乏临床研究,后来发现了更有效的小分子物质。与第一代ADC相比,第二代ADC具有更好的CMC特性。从当时FDA批准的三种二代ADC药物来看(vedotin、emtansine、ozogamicin),第二代ADC药物显示出良好的临床疗效和安全性。

rastuzumab emtansine(Kadcyla)结构式
然而,由于脱靶毒性、存在未结合抗体以及药物抗体比(DAR)为8引起的ADC聚集或快速清除等原因,目前大多数第二代 ADC显示出较窄的治疗窗口。此外,DAR>4的ADC被证明耐受性低、体内疗效低但是血浆清除率高。至此,二代ADC药物也难以满足患者的需求。优化单克隆抗体、连接子和结合毒性有效载荷可以提高第三代ADC的疗效,位点特异性偶联现在被认为是ADC成功开发的关键。
第三代抗体偶联药物
第三代ADC药物综合了一代二代失败的因素,利用小分子药物与单克隆抗体的位点特异性结合,产生DARs为2或4的ADC,这种ADC药物毒性降低,无未结合的单克隆抗体,稳定性和药代动力学大大提高,偶联脱落速度更低,药物活性高,低抗原水平下的细胞活性高。综上所述,第三代ADC药物攻克了导致一代二代药物失败的因素,让患者得到更好的治疗。
ADC的药代动力学特征
一般来说,在给药后,体内涉及四个过程。这些过程是吸收、分布、代谢和清除。
吸收
大多数抗体通常通过静脉注射或输液途径给予,抗体也可以通过皮下(SC)途径给予。然而,对于ADC,目前给药途径是静脉注射或输液。由于对细胞毒性有效载荷的反应和细胞毒性物质的局部沉积,SC给药可能不适用于ADC。
分布
药物在体内的分布可以用分布容积来描述。由于其大小和极性,抗体和ADC的分布通常局限于血管和间质间隙。
ADCs的初始分布一般局限于血管,其分布容积一般等于血容量。随后,ADCs可以分布到间质间隙。此外,ADC分布也会受到靶抗原表达和内吞的影响。
ADC在同一组织中的分布和积累会产生不良的(毒性)药理学影响,这是由于ADC的摄取后的细胞毒性药物或代谢物的释放。
代谢
ADC体内分解/代谢过程包括抗体分解代谢过程和小分子药物体内代谢。ADCs在到达肿瘤细胞前,在细胞内(non-cleavable linker)或者循环系统中(cleavable linker)释放效应分子,未结合的抗体和抗体片段遵循抗体的代谢途径通过酶解产生氨基酸,被机体重新利用。
ADC裂解或被分解代谢后可能形成的游离的小分子药物和/或连有氨基酸残基的小分子药物和/或linker的小分子药物代谢物,会进一步经历肝CYP450酶代谢,还可能发生潜在的药物药物相互作用。
除了ADC本身性质外,抗原的表达、受体/细胞密度,FcRn介导的循环作用、与Fcγ作用、受体介导的内吞作用、免疫原性等都会影响ADC的分解代谢。
清除
ADC也是通过分解代谢和排泄的方式进行消除。ADC可通过与靶点结合的特异途径,进入溶酶体后发生降解,释放小分子药物后从体内清除;还可以通过非特异的胞饮作用进行清除,该途径涉及新生儿受体(FcRn)参与的循环再利用过程。
ADC、抗体、分子量较大的多肽及氨基酸片段无法通过肾小球滤过排泄,而是以氨基酸的形式重新吸收利用。游离小分子药物、分子量较小的多肽及氨基酸连接的小分子药物、分子量较小的抗体片段可通过肾小球滤过进行排泄。同时,小分子药物及代谢产物也可经酶代谢消除或通过转运体排泄至粪便中。
ADC的生物分析
ADC有几种组分,为了表征这些组分的PK特征,需要几种分析方法,如下所述:
ELISA免疫分析测定结合物和总抗体的动力学曲线;
TFC-MS/MS,对游离药物/代谢物进行定量;
高分辨质谱用于体内药物抗体比(DAR)分析。
此外,两种类型的ELISA免疫分析用于定量测量ADC的分析物:第一种类型的分析测量总抗体,即DAR大于或等于零的ADC。第二种分析方法测量药物结合抗体,定义为DAR大于或等于1的ADC。
其它分析方法有尺寸排阻色谱法(SEC)和疏水作用色谱法(HIC)。SEC是最常用的液相色谱(LC)技术,用于测定抗体的聚集数量,该技术也可用于ADC。虽然HIC是一种用于蛋白质分离、纯化和表征的传统技术,但是这种技术现在正被用于ADC表征和分析。
细胞毒性有效载荷
ADC细胞毒性有效载荷应具备以下特性:
具有细胞毒性的有效载荷应具有恰当的脂溶性。
有效载荷的靶标应位于细胞内部。
有效载荷的分子应该是小尺寸的,缺乏免疫原性,可溶于水缓冲液,以便可以很容易地偶联。
有效载荷在血液中应该是稳定的。
目前,常用的细胞毒性药物效应分子为微管抑制剂(如:auristatins、maytansinoids)、DNA损伤剂(如calicheamicin、duocarmycins、anthracyclines、pyrrolobenzodiazepine dimers)和DNA转录抑制剂(Amatoxin和Quinolinealkaloid (SN-38))。已经获批上市的几个ADC药物共使用了6个不同的小分子药物,其中有3个ADC药物使用MMAE作为偶联药物,2个药物使用Calicheamicin作为偶联药物,另外成功应用的还有MMAF,DM1,SN-38,Dxd。
药物抗体比(DAR)
药物抗体比(DAR)是指附着在单个单抗上的有效载荷分子的平均数量,通常在2到4个分子之间。在极少数情况下,通过使用亲水链接器有效载荷可以安全地实现高达8的DAR,如Enhertus和Trodelvys。DAR对ADCs疗效的测定非常重要,此外,DAR可能影响药物在循环中的稳定性、PK和ADC的毒性。
研究表明,与DAR值<6的ADCs相比,DAR值高(7到14)的ADCs清除速度更快,体内疗效降低。DAR值及其对稳定性和PK的影响也取决于偶联位置和接头的大小。
赖氨酸或半胱氨酸通常被修饰以产生ADC。赖氨酸是连接底物和抗体的最常用的氨基酸残基之一, 赖氨酸通常存在于抗体表面, 因此容易偶联。Mylotargs、Kadcylas和Besponsas都使用赖氨酸生物结合技术。
其他氨基酸如半胱氨酸和酪氨酸也可以修饰,用马来酰亚胺修饰半胱氨酸合成了Adcetriss、Polivys、Padcevs、Enhertus、Trodelvys和Blenreps等ADC。
连接子
连接子(linker)是ADC不可或缺的一部分, 它决定ADC的药物释放机制、PK、治疗指数和安全性。早期的ADC连接体是化学不稳定的,如二硫化物和腙。这些连接体在循环中不稳定,半衰期短,一般为一到两天。最新一代的连接体在体循环中更稳定,如肽和葡萄糖醛酸连接体。两个最常见的连接体如下:
可裂解连接子
裂解型linker对细胞内环境敏感,在细胞内通过分解代谢和解离共同作用释放出游离的效应分子和抗体,如酸裂解连接体和蛋白酶裂解连接体。它们通常在血液中稳定,但在低pH和富含蛋白酶的溶酶体环境中会快速裂解,释放效应分子。此外,如果效应分子可以跨膜,则可通过发挥潜在的旁观者效应消灭肿瘤。
不可裂解连接子
不可裂解的linker是一种新一代的连接体,与可裂解的连接体相比,它具有更好的血浆稳定性。由于不可裂解的连接体可以提供比可裂解连接体更大的稳定性和耐受性,因此,这些连接体降低了靶外毒性,也提供了更大的治疗窗口。
免疫原性
在针对8个ADC的11个临床试验中, ADAs的基线发生率在1.4%到8.1%之间,基线后ADAs的发生率在0-35.8%之间,这些数值在治疗性单克隆抗体的范围内。总的来说,ADCs的ADA发生率在靶向血液肿瘤的患者比靶向实体肿瘤的患者少;大多数ADA是针对ADC的单克隆抗体结构域的。此外,在大多数患者中,这些ADC的半抗原样结构并不比治疗性单克隆抗体产生更多的免疫应答风险。
ADC药代动力学模型
应用模型的方法可以将PK、药效和安全性数据进行整合,以满足不同阶段ADC药物研发的需求,如:靶点的选择、抗体的亲和性、linker的稳定性、动物到人的外推、剂量的选择和调整、E-R相关性研究(exposure-response relationships)、DDI研究等等。由于ADC具有多种清除途径(解离和分解代谢),以及存在多种分析物的复杂的PK特征,使得其动力学模型也较为复杂。
不同的模型具有不同的应用,如可采用二房室模型和PBPK模型可以用清除率、解离、代谢速率等参数描述ADC的稳定性特征。目前非房室模型、群体药代模型、基于机制的模型、基于生理的模型在ADC药物动力学研究中均有应用。
小结
尽管第一个ADC在20多年前就获得了FDA的首次批准,但制药行业必须经历一个漫长且乏味的学习过程,才能在市场和临床开发中获得一条稳定的ADC管线。尽管目前已有15种已批准的ADC,但是由于ADC的技术进步,该领域正在经历一次爆发,我们在有效载荷,连接子和偶联技术方面获得了多个突破,进一步加深了对ADC这种新型药物的全面认识。
在ADC药物的研发进程中,临床药理学起着非常重要的作用,通过不断发展的生物分析技术,深入全面地阐明ADC药物的PK/PD特征,对于推动研发出更加低毒高效的ADC药物至关重要。ADC药物也必将在肿瘤治疗领域展现出更加强大的优势。
参考文献:
1.The Chemistry Behind ADCs. Pharmaceuticals (Basel). 2021 May; 14(5): 442.2. Clinical Pharmacology of Antibody-Drug Conjugates. Antibodies (Basel). 2021 May21;10(2):20.
3. Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245
原文标题:小药说药新春第三谈:抗体偶联药物
郑重声明:文章仅代表原作者观点,不代表本站立场;如有侵权、违规,可直接反馈本站,我们将会作修改或删除处理。
相关阅读




猜你喜欢
-
Focus | 世界核药的发展-RDC药物的兴起
2023-01-28 -
小药说药新春第四谈:CAR-NK
2023-01-28 -
小药说药新春第五谈:mRNA疫苗
2023-01-28 -
AI生成新春祝福海报,AIGC从“炫技”走向日常
2023-01-20 -
【聚焦】间充质干细胞优势特征凸显 我国相关药物研发进程不断加快
2022-12-19 -
盘点 | NAFLD/NASH全球在研药物进展
2022-11-14 -
肿瘤免疫治疗中的抗体亚型
2022-11-09 -
3D生物打印癌症模型:从基础生物学到药物开发
2022-11-09 -
药物发现中的斑马鱼疾病模型
2022-07-28 -
ADC药物的免疫原性
2022-06-20 -
解密抗体依赖增强,来自登革热疫苗的惊人发现
2022-05-17 -
吃药也可“编程”?盘点全球11家3D打印药物企业
2022-05-13 -
从类器官到类器官芯片,药物研发需要新方法
2022-05-10 -
开启未来之窗:AI与药物研发的共舞
2022-05-10 -
中俄“新春之会”加码能源合作 “多点开花”贸易新格局正形成
2022-02-07